
Development Regulatory Affairs & Pharmaceutical Regulation Q&A
- We are planning a consultation meeting with the PMDA but lack experience and are short-staffed. Could you advise us on how best to proceed and what points we should be attentive to?
It is crucial to objectively clarify the profile of the product under development based on the data obtained and consider what questions you want to ask during the consultation meeting. Understanding whether you are receiving essential advice from the PMDA to advance development, and preparing for the consultation meeting with the overall story in mind is important. Our company will fully support you in creating this narrative with our consultants who are experienced in negotiations.
- How long does it take to obtain a license after applying for a pharmaceutical manufacturing and marketing license?
The duration varies by prefecture, but if there are no issues with the application, it typically takes about 30 to 60 days.
- What is needed to obtain a manufacturing and marketing license for a pharmaceutical product?
There are two types of licenses: Type 1 for prescription pharmaceuticals and Type 2 for non-prescription pharmaceuticals. Applications must be submitted to the prefectural government to obtain these licenses. The specific application forms and materials required are detailed on each prefecture’s website.
- When is change management required after obtaining approval?
Changes in approved items for pharmaceuticals typically include the generic name, trade name, ingredients and amounts, manufacturing method, dosage and administration, efficacy and effects, storage methods and shelf life, specifications and test methods, manufacturing sites, and raw material manufacturing sites. The required documentation and procedures vary depending on the nature and extent of the changes.
- What requires approval for marketing authorization and what does not?
For pharmaceuticals, quasi-drugs, and cosmetics, Article 14 of the Pharmaceutical and Medical Device Act (PMD Act) requires that each product must obtain approval from the Minister of Health, Labour and Welfare for manufacturing and marketing. For medical devices and in vitro diagnostic pharmaceuticals, this requirement is outlined in Article 23-2-5 of the same act. There are exemptions to these rules, so for more detailed information, please consult with us.
CMC Regulatory Affairs Q&A
- What regulatory procedures are needed for changes that could affect the quality of the product, such as changes in the manufacturing method?
It is crucial to carefully consider what regulatory procedures are necessary for changes that could impact the product’s quality. We will thoroughly examine our client’s situation and propose appropriate solutions.
- There have been many incidents in post-marketing quality control in recent years. What are the causes?
Various cases are conceivable, including some leading to product recalls. A common cause is that the marketed products are not being manufactured and controlled appropriately according to the approval documents at the manufacturing facility.
- We are struggling with change management for overseas products. Can this be addressed?
Many companies face similar issues. Please consult with us as various cases can be considered.
- When and what needs to be done for foreign manufacturer accreditation?
Details on foreign manufacturer accreditation are provided on the PMDA website. As various cases are conceivable, please consult with us for specific guidance.
- Can a minor change be asked to be classified as a partial change by the PMDA?
Yes, if the PMDA finds the explanation for the scientific and rational basis insufficient, they may request a reclassification of the change.
Pharmacovigilance Q&A
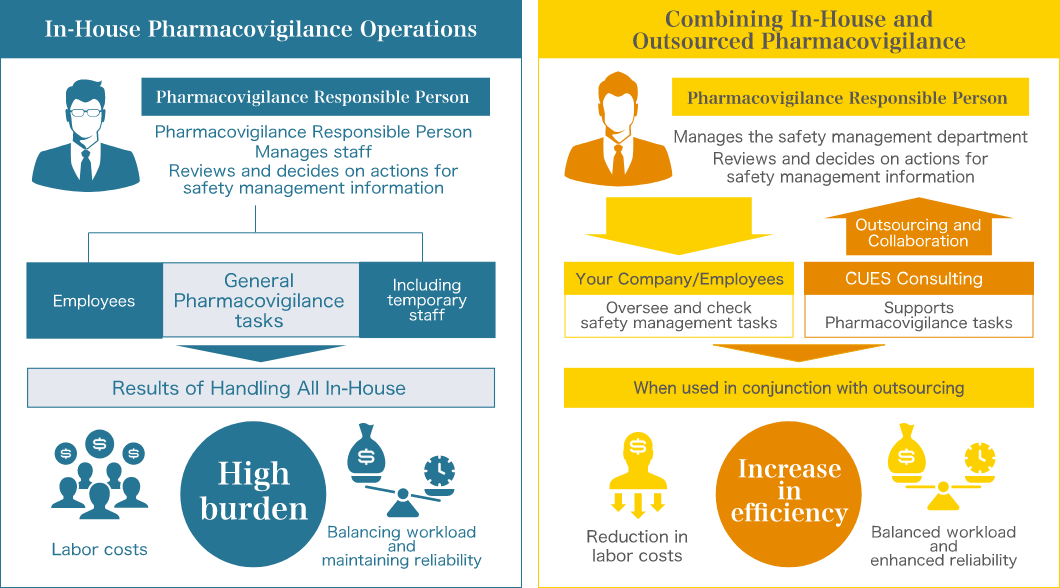
- What are the benefits of outsourcing Pharmacovigilance tasks?
Pharmacovigilance tasks are often urgent and can place a significant burden on staff, who may not always have the necessary expertise. By outsourcing routine tasks, operations can be maintained with minimal staff, reducing the burden and improving efficiency. Staff are also freed from tedious tasks, allowing them to focus on more advanced work.

- I'm unsure how to create an RMP. What should I do?
When creating a draft RMP for submission, many considerations need to be taken into account, such as evaluating the benefits and risks based on the product characteristics and information obtained during development, and considering overseas package insert drafts. Consult with us for guidance tailored to your specific situation.
- When is it a good time to introduce a safety database?
It is desirable to introduce it during the clinical trial phase, but even after the end of clinical trials, it’s recommended to introduce it before the product launch. If the product is post-launch and undergoing re-examination, introduction is advised to reconcile PMS data and for creating safety periodic reports, considering the reliability of the tasks and aligning with the current and future product portfolio.
- What are the benefits of using a safety database?
Using a safety database allows for automatic assignment of MedDRA and drug codes, significantly contributing to the reliability and efficiency of the work. Moreover, when creating periodic reports, data aggregation from the database greatly enhances the reliability of the document submission process to regulatory authorities.
General Services Q&A
- What is the process to request?
Please check the link below for more details.